
Software
Characterization of protein-protein interactions using AlphaFold
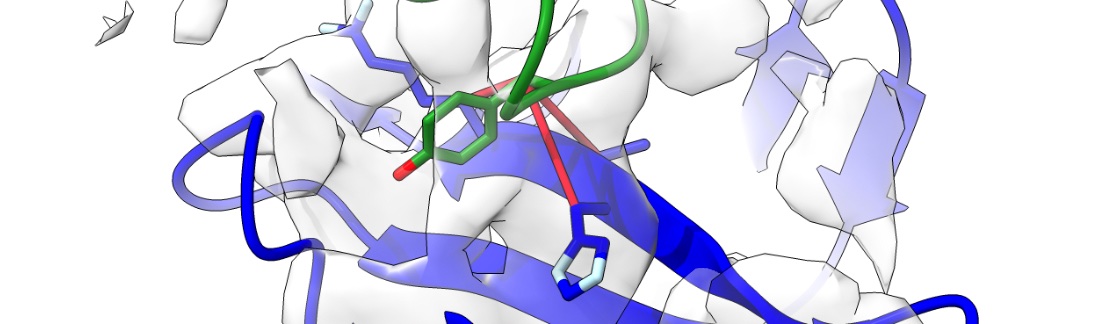
One of the hardest parts of macromolecular crystallography and cryo-EM is making sense of the structural models. We don’t use clay and wire to build them anymore, but the models have got bigger and the time pressure has got stronger over time. One of the problems associated with this - model validation - I have addressed with checkMySequence. However, there are cases where a model can be fitted into a map, but the resolution is too low to confirm its identity. Similarly, the resolution of the map may not allow the identification of contacts that are critical for functional analysis.
I have developed a tool based on AlphaFold2 - gapTrick - that helps to solve these problems. In principle, it rebuilds protein models, but it can also identify contacts at protein-protein complex interfaces. They are more reliable than confidence scores (pTM or ipTM) and very precise. Finally, gapTrick uses monomeric AlphaFold2 models and there is no training set bias that many AI-based tools suffer from!
I use it a lot for cryo-EM model building. Several fascinating stories will soon appear in the Structure stories section, stay tuned!
Sequence information identification and validation in EM and MX
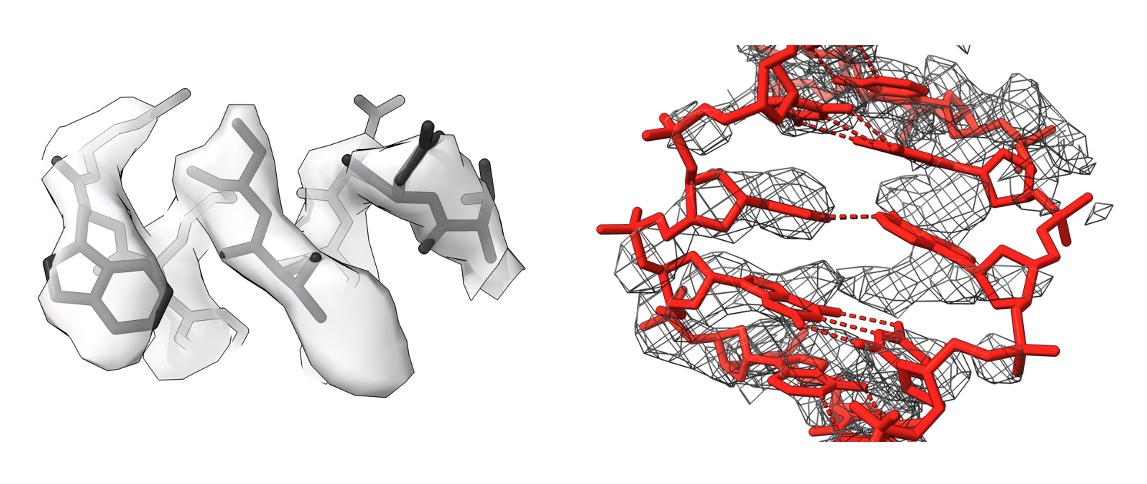
Sequence assignment is a critical step in macromolecular modelling in EM and MX, and often leads to hard-to-detect errors. The availability of AI-based predicted models hasn’t changed this. I have developed a suite of programs for the identification and validation of sequence information in models of macromolecules solved using EM and MX. The programs are avilable in CCP4, CCP4Cloud, CCPEM/doppio, and on https://gitlab.com/gchojnowski/
-
Chojnowski, “doubleHelix: nucleic acid sequence identification, assignment and validation tool for cryo-EM and crystal structure models” Nucleic Acids Research 2023. 51(15): p. 8255–8269
-
Chojnowski “Sequence-assignment validation in protein crystal structure models with checkMySequence.” Acta Cryst D (2023)
-
Chojnowski “Sequence-assignment validation in cryo-EM models with checkMySequence.” Acta Cryst D (2022): 806-816
-
Chojnowski et al. “findMySequence: a neural-network-based approach for identification of unknown proteins in X-ray crystallography and cryo-EM.” IUCrJ 9.1 (2022): 86-97
De novo model building in EM and MX
Before AlphaFold2 came along, ARP/wARP was one of the leading programs for modelling macromolecular crystal structures. I have made several contributions to the suite, including adapting it for modelling EM models. The programs are avilable in CCP4 and CCP4Cloud,
-
Chojnowski et al. “The accuracy of protein models automatically built into cryo-EM maps with ARP/wARP.” Acta Cryst D (2021): 142-150
-
Chojnowski, Pereira, and Lamzin. “Sequence assignment for low-resolution modelling of protein crystal structures.” Acta Cryst D (2019): 753-763
-
Chojnowski et al. “The use of local structural similarity of distant homologues for crystallographic model building from a molecular-replacement solution.” Acta Cryst D (2020): 248-260
Fragment based modeling of RNA structures
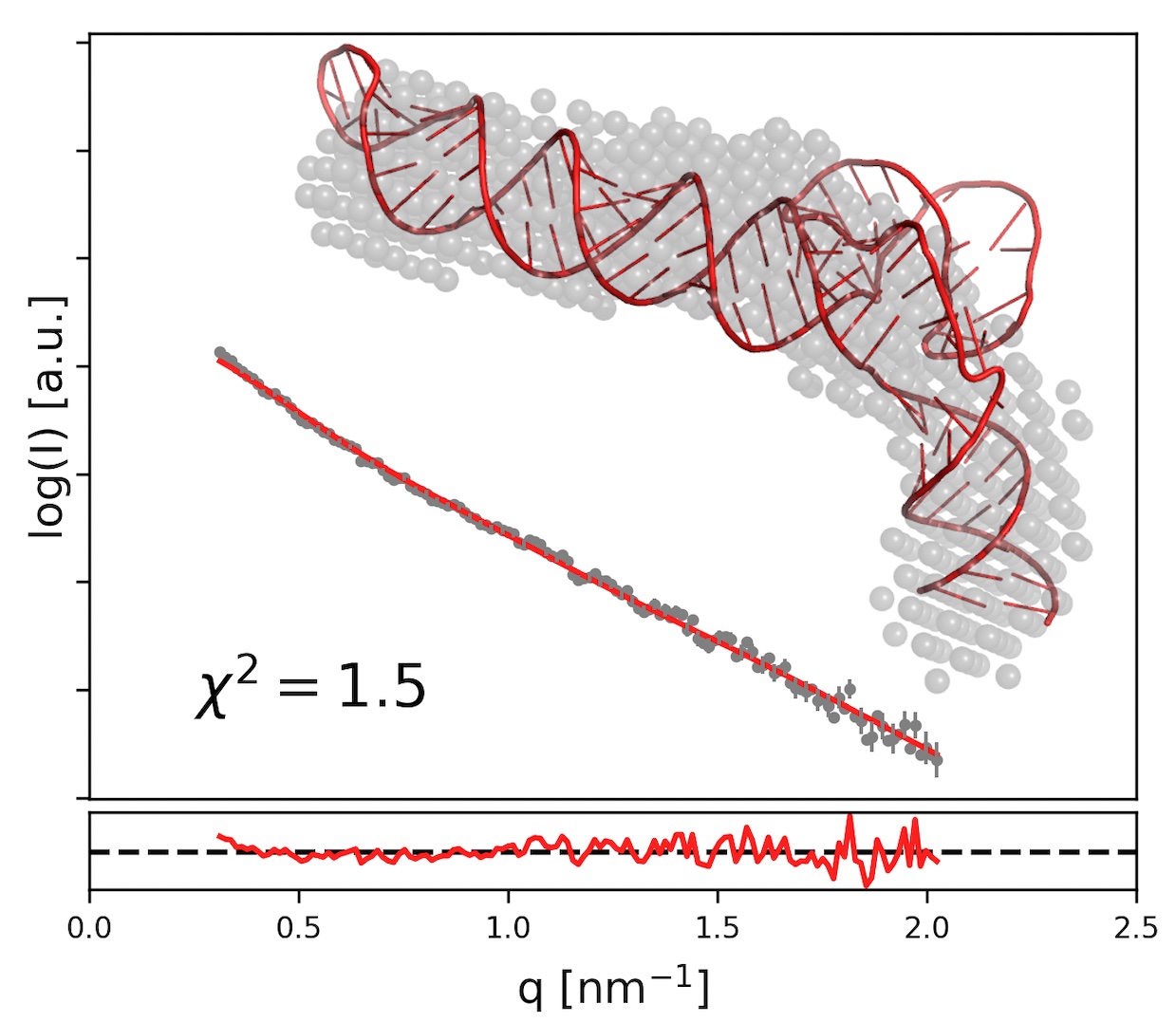
Structured RNA molecules often share structural motifs. I have developed a suite of programs for analysing existing RNA models (RNA Bricks database) and reassembling them with experimental restraints (BrickworX, RNAMasonry).
On the left is an example of a model of a viral RNA molecule built with RNA Masonry using experimental Small Angle X-ray Scatering (SAXS) data. RNAMasonry assembles models of RNAs from large, recurrent framents, signifficantly reducing the number of degrees of freedom. This is particularly important when building a 3D model of hundreds of nucleic acid residues from a 1D experimental SAXS curve with only a few independent data points.
-
Chojnowski et al. “Brickworx builds recurrent RNA and DNA structural motifs into medium-and low-resolution electron-density maps.” Acta Cryst D (2015): 697-705
-
Chojnowski, Waleń, and Bujnicki. “RNA Bricks—a database of RNA 3D motifs and their interactions.” Nucleic acids research 42.D1 (2014): D123-D131
-
Chojnowski et al. “RNA 3D structure modeling by fragment assembly with small-angle X-ray scattering restraints.” Bioinformatics 39.9 (2023): btad527
Unusual signals in protein crystal diffraction patterns
Series of publications from my PhD on the identification of unusually strong signals in protein crystal diffraction patterns using statistical analysis based on the Extreme Value Distribution Theorem. The initial idea was to see if one could find signals associated with secondary structure elements (e.g. alpha helices) and use them for molecular replacement. However, these turned out to be too weak to be able to pick out the poseudo-random fluctuations. I used this idea to write a program called DIBER, which identifies RNA/DNA helices based on diffraction patterns alone, before the crystal structure is solved.
-
Chojnowski, Bujnicki, and Bochtler. “RIBER/DIBER: a software suite for crystal content analysis in the studies of protein–nucleic acid complexes.” Bioinformatics 28.6 (2012): 880-881
-
Chojnowski and Bochtler. “DIBER: protein, DNA or both?.” Acta Cryst D (2010): 643-653
-
Chojnowski and Bochtler. “The statistics of the highest E value.” Acta Cryst A (2007): 297-305
-
Bochtler and Chojnowski. “The highest reflection intensity in a resolution shell.” Acta Cryst A (2007): 146-155